

Introducing PGT-H: A New Form of Embryo Screening for Couples with Shared Ancestry
With PGT-H, couples can reduce their children's risk of congenital anomalies, intellectual disability, and infant death.

The Herasight team is proud to introduce preimplantation genetic testing for homozygosity (PGT-H), a new screening approach designed for consanguineous couples who share a recent common ancestor. When parents are related, their children are more likely to inherit identical copies of the same chromosomal segments from both sides of the family, a state known as homozygosity. Extended regions of homozygosity increase the chance that harmful recessive variants are present in both copies of a gene, substantially elevating disease risk. PGT-H targets this risk directly, using data already collected during routine IVF to rank embryos by their level of genome-wide homozygosity. This helps reduce disease risk across a broad range of conditions simultaneously.
Table of contents
What is the impact of consanguinity?
Consanguinity is well established as a meaningful risk factor for a range of serious health outcomes. Children of first-cousin couples face roughly 2.5 times the population risk of intellectual disability (Jamra, 2018) and about twice the risk of both infant death (Stoltenberg et al., 1998) and major congenital anomalies (Sheridan et al., 2013). When parents are at risk of passing on two copies of a known recessive variant, this risk can already be reduced through preimplantation genetic testing for monogenic disorders (PGT-M), which is increasingly used during IVF in some regions with high rates of consanguinity. However, PGT-M is limited to variants that have already been identified and classified as pathogenic. Many harmful recessive variants in consanguineous families are not yet recognized as disease-causing, and are therefore missed by current screening methods. PGT-H goes a step further by reducing the fraction of the genome in runs of homozygosity (FROH), thereby lowering recessive disease risk beyond known variants alone.
Consanguineous relationships are typically among second cousins, first cousins, and double first cousins (Figure 1B). Second cousins share one pair of great-grandparents. First cousins share one pair of grandparents, meaning their parents are siblings. Double first cousins are when two siblings from one family each have children with two siblings from another family, sharing both sets of grandparents. These relationships yield progressively higher expected offspring homozygosity, with average FROH (fraction of the genome in runs of homozygosity) values of approximately 1.6%, 6.25%, and 12.5% respectively. First-cousin and closer unions are particularly prevalent across West Africa through the Middle East and into South and Central Asia, where between 20% and 50% of marriages are between second cousins or closer (Figure 1A adapted from Hamamy et al., 2011).
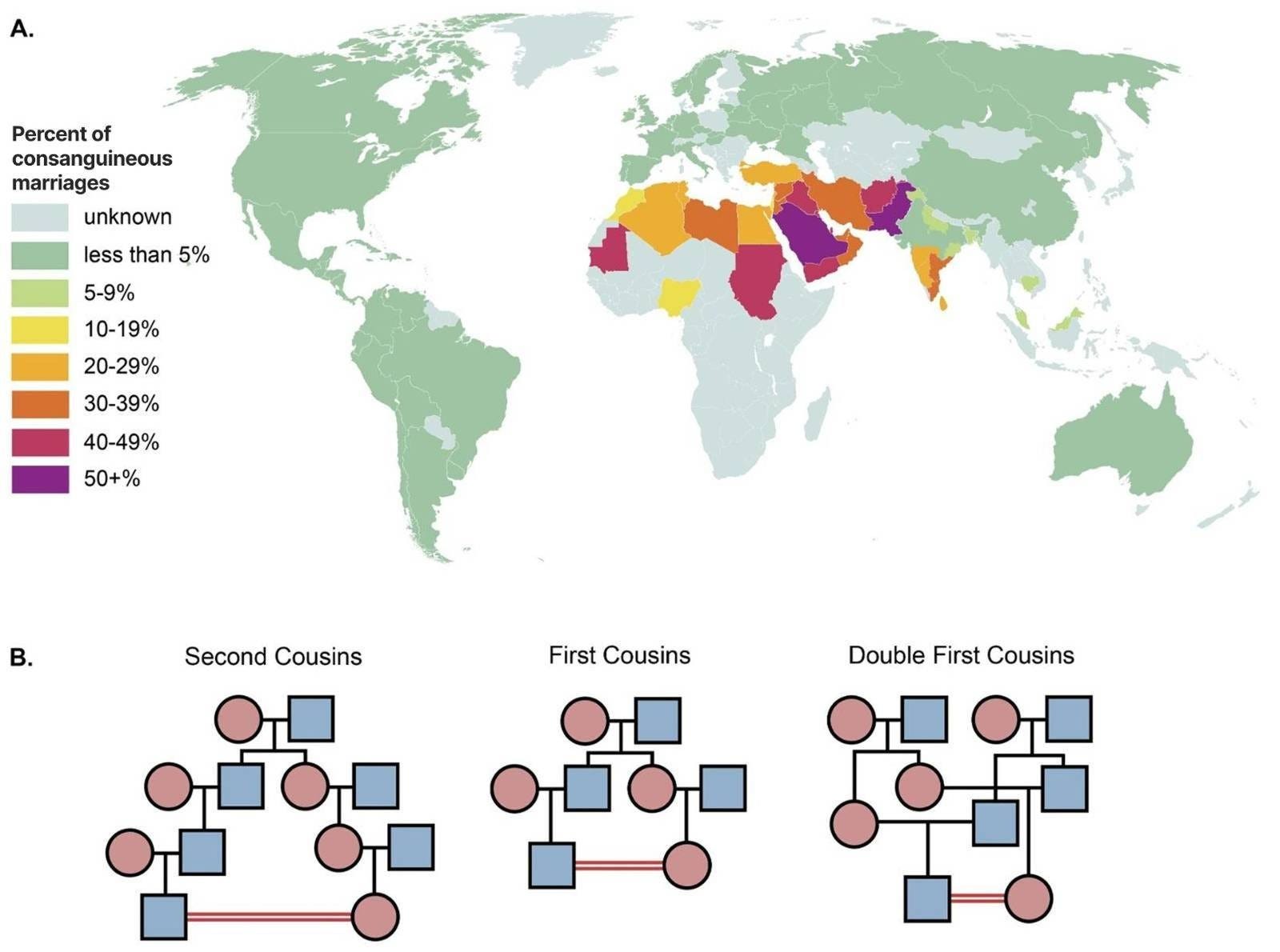
The health consequences of consanguinity are well-documented and span a wide range of adverse outcomes. Children of related parents face higher risks of stillbirth, infant death, congenital anomalies, developmental delay, and intellectual disability (Zlotogora & Shalev, 2010; Stoltenberg et al., 1998; Small et al., 2024; Sheridan et al., 2013). A Norwegian registry study spanning over two decades found that consanguinity was associated with elevated rates of stillbirth and infant mortality even after adjusting for maternal education and other covariates (Stoltenberg et al., 1998). Children of first-cousin parents have also been shown to have elevated risks of early-life mortality, higher healthcare utilization, and increased rates of speech, language, and learning difficulties (Small et al., 2024).
Using published baseline risks and published estimates of how risk rises with parental relatedness, we estimate that infant mortality rises from about 0.6% in the outbred population to about 1.3% in children of first cousins and about 2.0% in children of double first cousins (Ely & Driscoll, 2025; Stoltenberg et al., 1998). Risk of intellectual disability rises from about 2% to about 5% and 8% respectively (Jansen et al., 2023; Jamra, 2018), while risk of major congenital anomalies rises from about 3% to about 6.6% and 10.2% (Centers for Disease Control and Prevention, 2008; Sheridan et al., 2013). These estimates are likely conservative, as real populations with historically elevated consanguinity typically carry background FROH of 1–3%, which would widen the distribution of autozygosity among embryos and increase absolute risk further.
The effects extend beyond classic recessive disease. Higher genome-wide autozygosity has been linked to a broad range of common conditions, including cardiometabolic disease, higher BMI, reduced cognitive performance, lower fertility, and increased risk of type 2 diabetes (Ceballos et al., 2018; Clark et al., 2019; Ceballos et al., 2020; Malawsky et al., 2023). In our model, lifetime type 2 diabetes risk rises from about 20% in the outbred population to about 28% in children of first cousins and about 38% in children of double first cousins (Narayan et al., 2007; Malawsky et al., 2023). Elevated autozygosity is also associated with higher rates of childlessness, with lifetime risk rising from about 20% in the outbred population to about 34% in children of first cousins and about 51% in children of double first cousins (Guzzo & Loo, 2023; Clark et al., 2019). Much of this broad burden is likely driven by recessive risk distributed across the genome rather than by a small number of known pathogenic variants, which is precisely what monogenic testing cannot address.
Why embryos from the same couple differ
Embryos from the same couple are not genetically identical. Each embryo inherits a different combination of chromosomal segments from the mother and father, because chromosomes are shuffled during the formation of eggs and sperm through a process called recombination, and then passed on at random. The location and extent of these recombination events differs for every egg and every sperm, meaning that no two embryos inherit exactly the same patchwork of parental DNA.
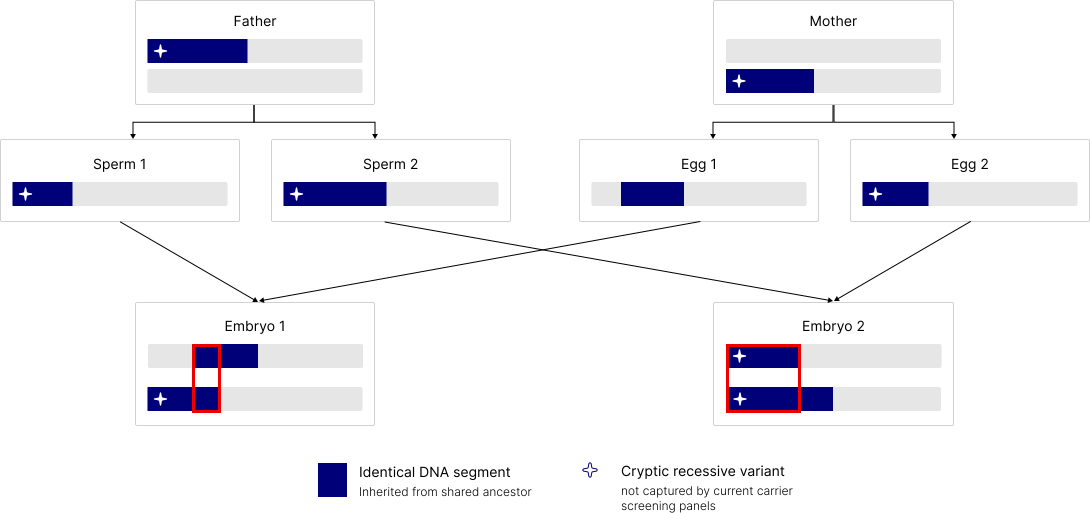
This variation also affects autozygosity. Figure 2 illustrates the mechanism: both parents carry an identical DNA segment inherited from their shared ancestor, marked in blue. When that segment happens to be passed down through both the sperm and the egg to the same embryo, the cryptic recessive variant it contains becomes homozygous. Importantly, this variant is not captured by standard carrier screening panels. The second embryo in the figure inherits a different combination of segments and carries a different autozygosity pattern entirely. Both embryos come from the same parents, but their recessive disease risk differs.
To quantify how much this variation matters in practice, we performed forward-time simulations of consanguineous pedigrees using realistic recombination maps (Bhérer et al., 2017), generating 1,000 embryos across 1,000 independent first-cousin and double-first-cousin families. As expected, the mean FROH across embryos clustered near the theoretical values of 6.25% for first-cousin families and 12.5% for double-first-cousin families. But individual embryos within the same family spanned a broad range around those means, with some embryos falling well below and others well above the family average (Figure 3).
This within-family variation is what makes PGT-H possible. Even when the degree of parental relatedness is fixed, the recessive disease risk is not the same across all embryos from that couple. Some embryos inherit a lower-autozygosity combination of segments by chance, and those embryos carry meaningfully lower expected recessive disease risk. PGT-H identifies and prioritizes those embryos.
What our simulations show
For first-cousin couples, selecting the lowest-FROH embryo from a cohort of five reduces FROH by approximately 40% on average relative to a randomly selected embryo. Translating this into disease risk, PGT-H reduces intellectual disability risk from approximately 5% to 2.8–3.2%, approaching the outbred population baseline of around 2%. Risk of major congenital anomalies falls from 6.6% to 4.0–4.5%, infant death risk falls from 1.3% to around 1.0%, and type 2 diabetes risk falls from 28% to around 24.5%. For double-first-cousin couples the reductions are larger in absolute terms: intellectual disability risk falls from 8% to 4.1–4.9%, and congenital anomaly risk falls from 10.2% to 5.5–6.7%. These estimates are conservative, as real populations with historically elevated consanguinity typically carry background FROH of 1–3%, which would widen within-family variance and increase the achievable benefit further (Clark et al., 2019).
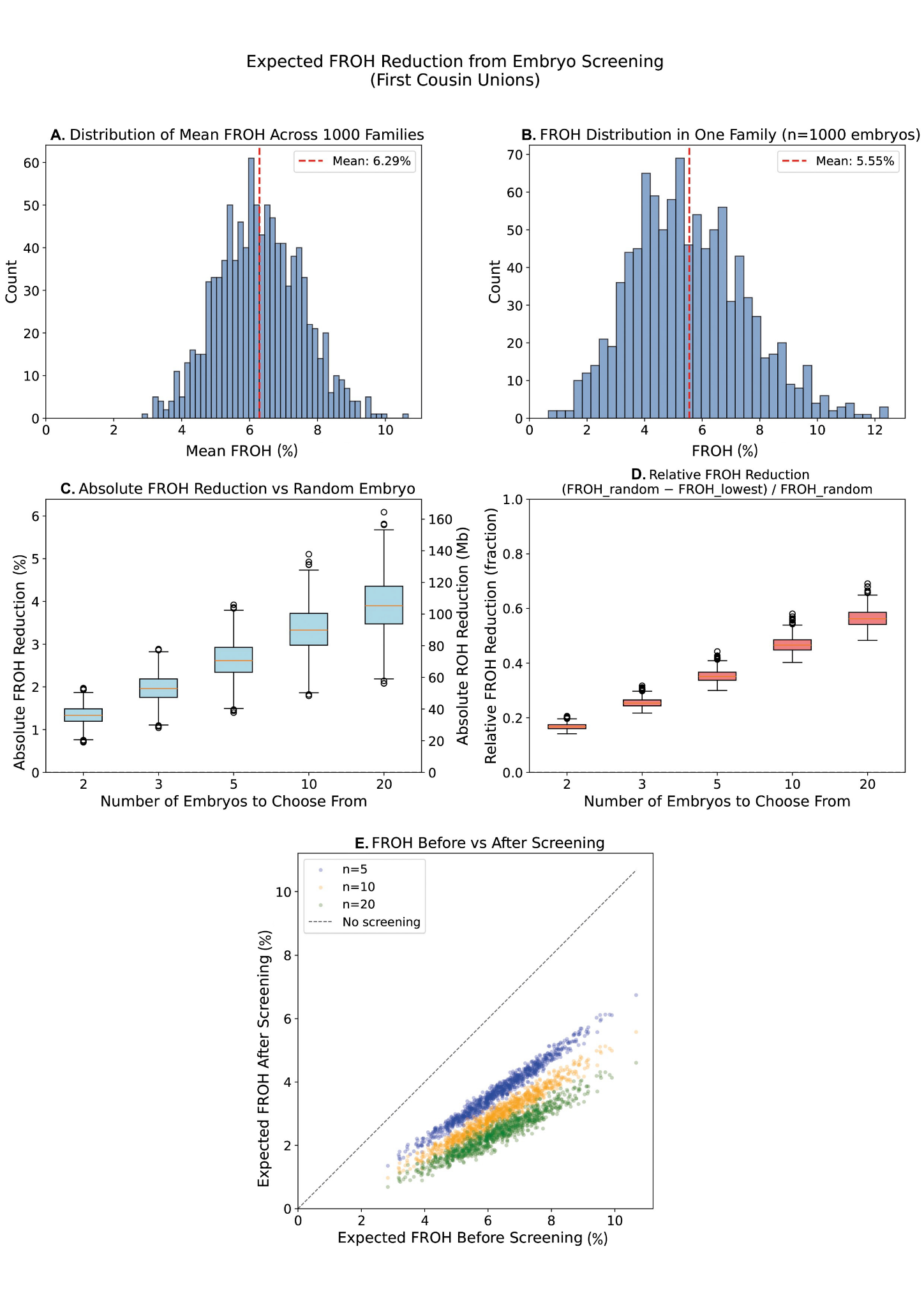
To understand where these numbers come from, it helps to walk through our simulations in detail. We simulated 1,000 independent first-cousin families and generated 1,000 embryos per family using realistic sex-specific recombination maps (Bhérer et al., 2017). Panel A of Figure 3 shows the distribution of mean FROH across those 1,000 families, clustered near the theoretical expectation of 6.25% for first-cousin unions. Panel B zooms into a single representative family and shows the distribution of FROH across its 1,000 simulated embryos. The spread is substantial: embryos from the same two parents range from close to zero up to well above 10% autozygosity. The same couple can produce embryos that differ by several percentage points in their genome-wide homozygosity purely by chance.
Panels C and D show what this variation means for screening. Panel C plots the expected absolute reduction in FROH when selecting the lowest-FROH embryo from cohorts of 2, 3, 5, 10, and 20 embryos. With five embryos, the average absolute reduction is roughly 3 percentage points for first-cousin couples, corresponding to a relative reduction of approximately 40% shown in Panel D. The benefit grows steadily with cohort size, though with diminishing returns. Panel E puts this in practical terms: each point represents a simulated family, with the x-axis showing expected FROH without screening and the y-axis showing expected FROH after selecting the lowest-FROH embryo. Across the full range of families, screening consistently shifts embryos below the diagonal, meaning essentially every family benefits regardless of where their mean FROH happens to fall.
The simulation results are also supported by a mathematical model derived from first principles. The expected reduction in FROH from selecting the best embryo among n candidates scales approximately with the square root of the logarithm of n, weighted by the within-family standard deviation of FROH. For first-cousin couples this gives an expected FROH reduction of approximately 1.73% times the square root of the natural log of n, a result that closely matches the simulated reductions and confirms that the gains from embryo selection are both real and predictable. A feature of PGT-H that distinguishes it from polygenic embryo screening is that by reducing genome-wide autozygosity, a single embryo ranking simultaneously lowers risk across all autozygosity-associated outcomes at once. The reductions in infant death, intellectual disability, congenital anomalies, type 2 diabetes, and childlessness are all achieved through the same ranking, without any tradeoff between them.
What this looks like in practice
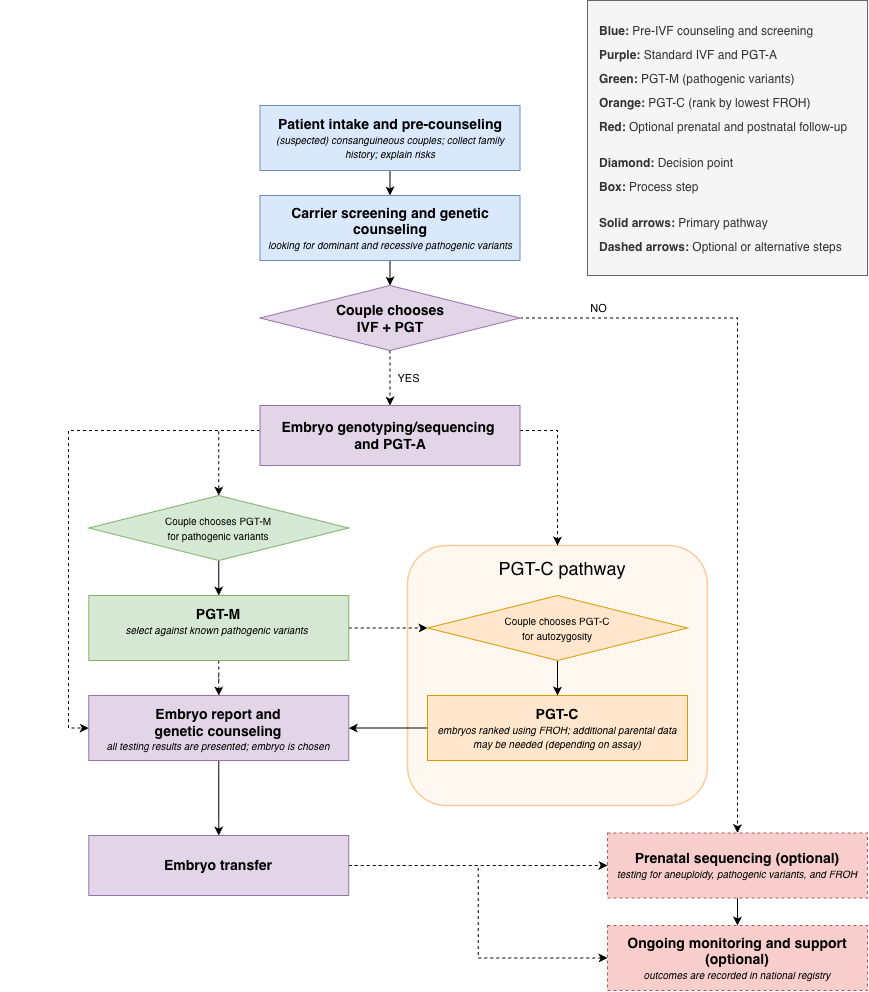
In practice, PGT-H would begin with couples who already know they are related, such as first or second cousins, as well as couples from founder, bottlenecked, or highly endogamous populations where recessive risk may be elevated even without a known recent relationship. These couples would first undergo genetic counseling and parental sequencing to measure shared ancestry directly in the genome and assess the degree of consanguinity. If that level were high enough to raise concern, they could choose to pursue IVF with embryo screening.
The first step within an IVF cycle would be addressing known pathogenic variants. Carrier screening and parental sequencing can identify shared recessive variants, as well as relevant dominant pathogenic variants, that can be screened against directly using PGT-M. Embryos would also undergo standard PGT-A to screen for chromosomal abnormalities. This means that known genetic risks are addressed first, using the same variant-based approaches already familiar in clinical IVF.
PGT-H would often serve as a final embryo prioritization step after PGT-A and PGT-M. Rather than treating all embryos from the same couple as genetically equivalent, PGT-H uses the embryo-level FROH as an additional decision factor, prioritizing embryos with lower autozygosity and therefore lower expected recessive disease risk across the genome. Importantly, PGT-H does not require any additional embryo biopsy beyond what is already performed for PGT-A. Parental whole genome sequencing in combination with the ultra-low-pass sequencing already collected during PGT-A is sufficient to reconstruct embryo-level FROH with high fidelity (Li et al., 2025). The full workflow is illustrated in Figure 4. In this way, PGT-H complements rather than replaces existing embryo screening, adding a genome-wide layer of recessive risk reduction that operates entirely beyond the reach of variant-level approaches.
Conclusion
Existing approaches address known variants but leave the large majority of genome-wide recessive risk for consanguineous couples untouched. PGT-H fills that gap by targeting autozygosity directly, using data already generated in routine IVF workflows and requiring no additional procedures. Our simulations show that selecting the lowest-FROH embryo from a typical cohort meaningfully reduces risk across a broad spectrum of serious outcomes simultaneously, from intellectual disability and congenital anomalies to type 2 diabetes and infant death. Combined with PGT-M, the two approaches act in a complementary fashion, addressing different components of the same underlying problem.
We anticipate that PGT-H will be most impactful in the regions where consanguinity is most common, many of which already face barriers to advanced reproductive genomics. Ensuring access will require investment in insurance or national health coverage, culturally sensitive genetic counseling, and clinical infrastructure in those settings. We are proud to offer PGT-H as a meaningful step toward reducing the burden of recessive disease for consanguineous families worldwide.
—
For full technical details, please see our PGT-H preprint. If you are interested in PGT-H for your IVF cycle, sign up here. Subscribe to our newsletter for more updates.
References
Bhérer, C., Campbell, C. L., & Auton, A. (2017). Refined genetic maps reveal sexual dimorphism in human meiotic recombination at multiple scales. Nature Communications, 8, 14994. https://doi.org/10.1038/ncomms14994
Ceballos, F. C., Joshi, P. K., Clark, D. W., Ramsay, M., & Wilson, J. F. (2018). Runs of homozygosity: Windows into population history and trait architecture. Nature Reviews Genetics, 19, 220–234. https://doi.org/10.1038/nrg.2017.109
Ceballos, F. C., Hazelhurst, S., Clark, D. W., et al. (2020). Autozygosity influences cardiometabolic disease-associated traits in the AWI-Gen Sub-Saharan African study. Nature Communications, 11, 5754. https://doi.org/10.1038/s41467-020-19595-y
Centers for Disease Control and Prevention. (2008). Update on overall prevalence of major birth defects — Atlanta, Georgia, 1978–2005. MMWR Morbidity and Mortality Weekly Report, 57, 1–5. https://www.cdc.gov/mmwr/preview/mmwrhtml/mm5701a2.htm
Clark, D. W., Okada, Y., Moore, K. H. S., et al. (2019). Associations of autozygosity with a broad range of human phenotypes. Nature Communications, 10, 4957. https://doi.org/10.1038/s41467-019-12283-6
Ely, D., & Driscoll, A. (2025). Infant mortality in the United States, 2023: Data from the period linked birth/infant death file. National Center for Health Statistics. https://doi.org/10.15620/cdc/174592
Guzzo, K., & Loo, J. (2023). Number of children to women aged 40–44, 1980–2022. National Center for Family and Marriage Research. https://doi.org/10.25035/ncfmr/fp-23-29
Hamamy, H., Antonarakis, S. E., Cavalli-Sforza, L. L., et al. (2011). Consanguineous marriages, pearls and perils: Geneva International Consanguinity Workshop report. Genetics in Medicine, 13, 841–847. https://doi.org/10.1097/GIM.0b013e318217477f
Jamra, R. (2018). Genetics of autosomal recessive intellectual disability. Medizinische Genetik, 30, 323–327. https://doi.org/10.1007/s11825-018-0209-z
Jansen, S., Vissers, L. E. L. M., & De Vries, B. B. A. (2023). The genetics of intellectual disability. Brain Sciences, 13, 231. https://doi.org/10.3390/brainsci13020231
Li, J. H., Ahangari, M., Wolfram, T., & Christensen, M. (2025). ImputePGTA: Imputing embryo genomes from low-pass sequencing and whole-genome parental data. medRxiv. https://doi.org/10.1101/2025.11.07.25339763
Malawsky, D. S., Van Walree, E., Jacobs, B. M., et al. (2023). Influence of autozygosity on common disease risk across the phenotypic spectrum. Cell, 186, 4514–4527. https://doi.org/10.1016/j.cell.2023.08.028
Narayan, K. M. V., Boyle, J. P., Thompson, T. J., Gregg, E. W., & Williamson, D. F. (2007). Effect of BMI on lifetime risk for diabetes in the U.S. Diabetes Care, 30, 1562–1566. https://doi.org/10.2337/dc06-2544
Sheridan, E., Wright, J., Small, N., et al. (2013). Risk factors for congenital anomaly in a multiethnic birth cohort: An analysis of the Born in Bradford study. The Lancet, 382, 1350–1359. https://doi.org/10.1016/S0140-6736(13)61132-0
Small, N., Kelly, B., Malawsky, D. S., Lodh, R., Oddie, S., & Wright, J. (2024). Mortality, morbidity and educational outcomes in children of consanguineous parents in the Born in Bradford cohort. Wellcome Open Research, 9, 319. https://doi.org/10.12688/wellcomeopenres.22547.2
Stoltenberg, C., Magnus, P., Lie, R. T., Daltveit, A. K., & Irgens, L. M. (1998). Influence of consanguinity and maternal education on risk of stillbirth and infant death in Norway, 1967–1993. American Journal of Epidemiology, 148, 452–459. https://doi.org/10.1093/oxfordjournals.aje.a009670
Zlotogora, J., & Shalev, S. A. (2010). The consequences of consanguinity on the rates of malformations and major medical conditions at birth and in early childhood in inbred populations. American Journal of Medical Genetics Part A, 152A, 2023–2028. https://doi.org/10.1002/ajmg.a.33537
Uh-oh, I suspect people from cousin marriage countries who have enough foresight and ability to pay for IVF selection can afford to buy a wife who is not a cousin./s