

ImputePGTA
Bringing polygenic embryo prediction to IVF clinics worldwide

We are announcing ImputePGTA, a new technique that enables accurate polygenic embryo screening from a routine test (PGT-A) performed in IVF clinics around the world.
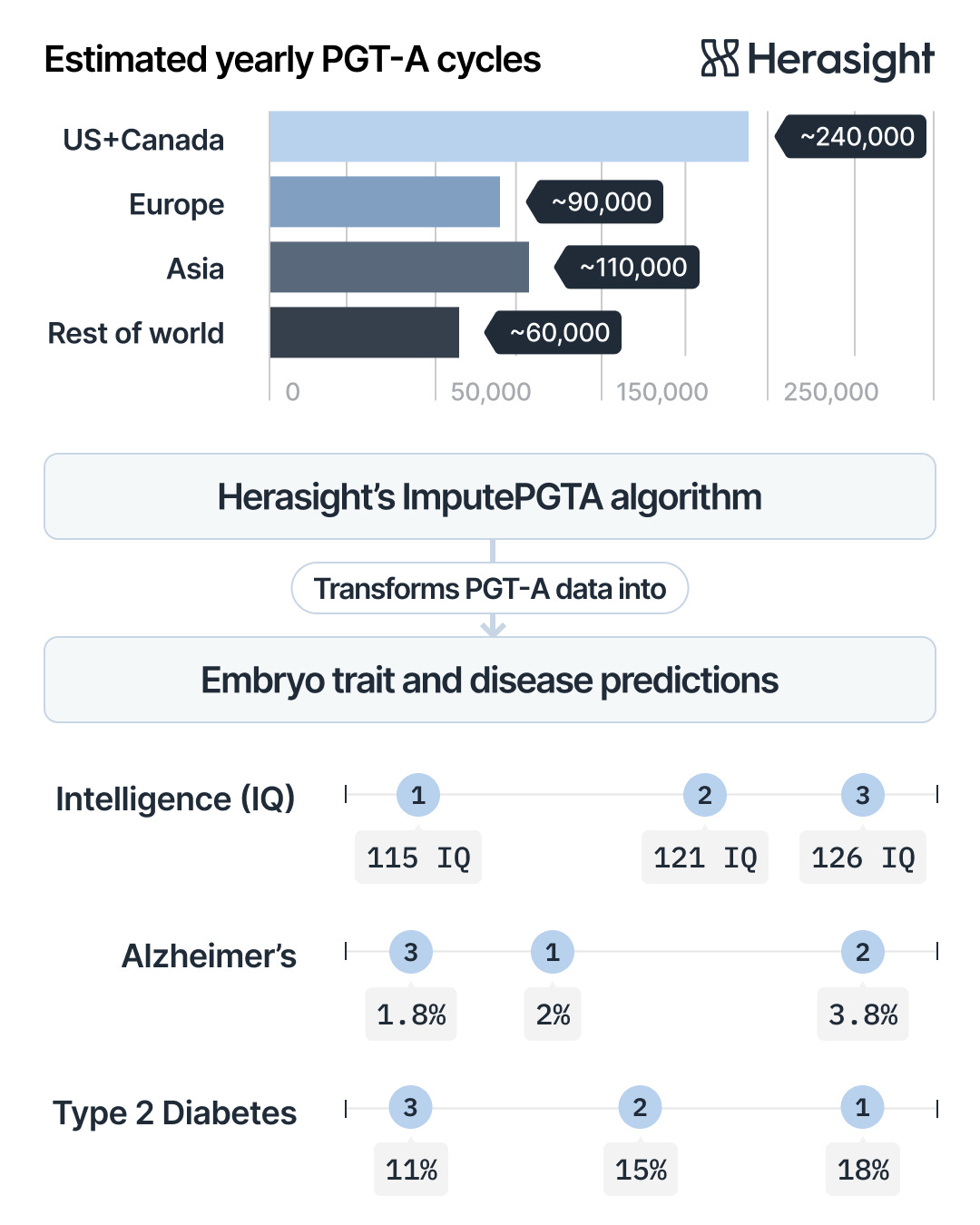
ImputePGTA is a revolutionary technique that enables us to combine whole-genome sequencing of parents with data derived from a routine test offered by IVF clinics (preimplantation genetic testing for aneuploidy, or PGT-A) to reconstruct the whole unique genome of each embryo.
ImputePGTA allows couples around the world to access Herasight’s world-class genetic predictors for IQ and many diseases through their local IVF clinic. This post summarizes the implications of ImputePGTA, as well as the science behind our whitepaper, “ImputePGTA: Accurate Embryo Genotyping and Polygenic Scoring from Ultra-Low-Pass Sequencing.”
Have you done IVF before, or are you thinking about it? Are you considering testing your embryos? Sign up here.
Table of Contents
Implications for parents using IVF
ImputePGTA is the culmination of years of dedicated research and was made possible through several important breakthroughs in genomic science. It enables accurate preimplantation genetic testing for polygenic traits (PGT-P) using embryo data already generated from PGT-A tests that are routinely performed around the world, removing the main practical barrier to widespread adoption of the technology.
Parents who wish to use polygenic scores to guide embryo screening have been limited by the difficulty of obtaining comprehensive genome data on their embryos, currently only available from two US providers. This is because it is much easier to whole-genome sequence an adult by collecting a tiny amount of blood or saliva than it is to sequence an embryo, from which only a few cells can be taken.
ImputePGTA changes the situation dramatically. PGT-A testing is offered at IVF clinics around the world, and parental genome sequencing is much easier than embryo sequencing. ImputePGTA allows us to combine sparse PGT-A data with comprehensive parental genome data — using both short- and long-read sequencing — to give us a full picture of the embryo’s genome without the need to use expensive and specialized wet-lab procedures that are not widely available.
ImputePGTA lowers a crucial barrier for parents to access information about polygenic diseases and traits. It is easy for couples using IVF to test for monogenic diseases (either through carrier screening before IVF, or through PGT-M after they create embryos). But monogenic diseases are rare. The vast majority of diseases that have a genetic component are polygenic — they are caused by many genes of small effect — and can arise even when there is no family history of the disease.
ImputePGTA will allow parents and IVF clinics to use polygenic screening without needing to partner with a lab that offers specialized embryo genotyping procedures. This means that Herasight integrates into the existing IVF process without adding additional burden on physicians: we help patients obtain the raw data from the PGT-A test, sequence the parents, and offer counseling from board-certified genetic counselors to help patients understand their embryo reports.
Background
Herasight offers advanced genetic screening, with one of our most important offerings being our best-in-class polygenic scores for predicting disease risks using PGT-P (preimplantation genetic test for polygenic conditions) (Moore et al., 2025). PGT-P uses polygenic scores to predict the traits and disease risks of embryos, enabling parents to select embryos according to these predictions.
Until now, one of the main practical barriers to performing PGT-P has been that specialized lab techniques were required to accurately capture genome-wide information needed for polygenic prediction (Xia et al., 2024; Treff et al. 2019). Currently, this means that embryo biopsies must be sent to specialized labs located in the US. Since the vast majority of IVF clinics and genetic testing providers are not equipped to perform these complex procedures, clinics had to partner with one of the very few existing PGT-P providers, severely limiting access to this new technology. This has thus far imposed significant operational overhead for those who wish to use PGT-P, and imposes geographical limitations on those abroad.
On the other hand, PGT-A, which screens for chromosomal abnormalities such as Down syndrome, is routinely used in ~60% of IVF cycles in the United States and is commonly available globally, with uptake rates varying due to varying regulatory environments (Munne et al. 2024, Smeenk et al., 2024; Smeenk et al., 2025).
Using data from standard PGT-A tests to analyze the embryo’s entire genome therefore dramatically reduces barriers to performing PGT-P. However, standard PGT-A techniques only generate extremely low-coverage genome sequencing data, typically around 0.002–0.004× depth (Heiser et al., 2023). While this ultra-low-pass (ULP sequencing is enough to detect large chromosomal issues, accurately identifying individual genetic variants usually requires much higher coverage. For comparison, high-quality whole-genome sequencing typically has about 30× coverage, meaning each position in the genome is read roughly 30 times. This is about 7,500 (!) times more data than the 0.004× coverage provided by standard PGT-A.
A simple solution to a hard problem
{kind=link}
The sparsity of PGT-A sequence data means that without additional information, reconstruction of the embryo’s whole genome is an intractable problem. In contrast to typical experimental setups in genomics, however, embryo genotyping in IVF benefits from the fact that parental genomes are routinely available for sequencing. This is important because from the laws of Mendelian inheritance, we know that an embryo’s genome is a mosaic of its parental genomes (with the exception of de novo mutations). If we are able to identify exactly which bits of the parental genomes were inherited, we would then be able to accurately reconstruct the inherited embryo genome without having to obtain direct measurements of each position in the embryo’s genome.
ImputePGTA leverages precisely this fact — by using knowledge of parental genomes and the ultra-sparse data generated for an embryo from PGT-A, it identifies the segments of each parental genome that were actually passed down to that embryo, and fills in the missing pieces using the known genetic data from the respective parent. In the following, we break down this process.
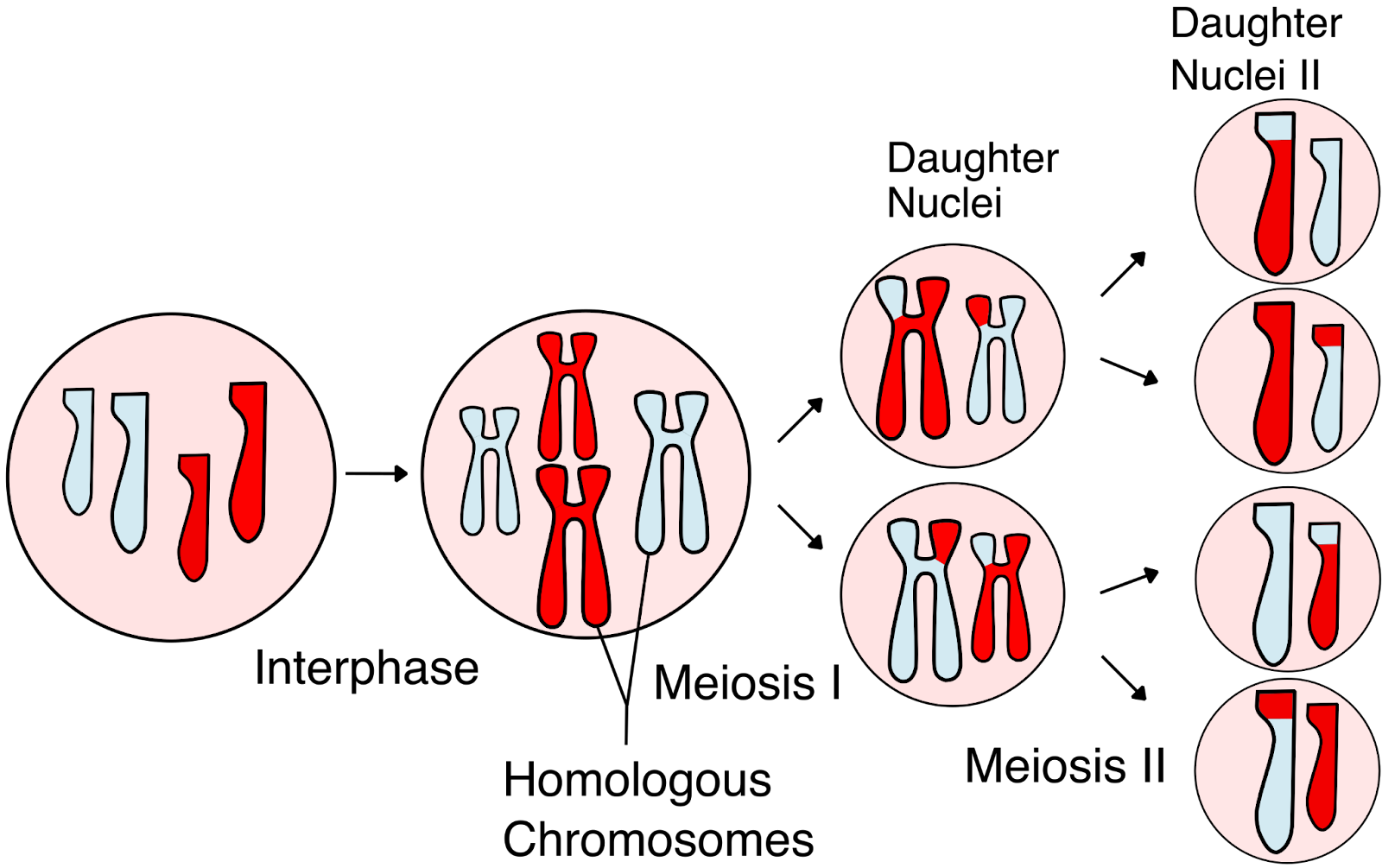
From the basic laws of Mendelian inheritance, we know that every embryo inherits one copy of each chromosome from each parent. Before this happens, each parent’s chromosomes are mixed through a process called meiotic recombination, in which DNA segments are exchanged between the two chromosome copies in each parent (Figure 1). This shuffling creates new combinations of genetic variants called haplotypes. Because of recombination, parents pass on unique mosaics of these haplotypes to each child. On average, several dozen recombination events occur in each generation, creating a distinct combination of parental DNA in every child’s chromosomes.
PGT-A data only provides a tiny sampling of the genome — often just a few scattered reads. But that sparse information can still tell us something important. If we can identify where the recombination breakpoints occurred — i.e., the boundaries of the segments that were actually inherited by the embryo — we should be able to effectively reconstruct the embryo’s genome by piecing together the correct combinations of maternal and paternal DNA.
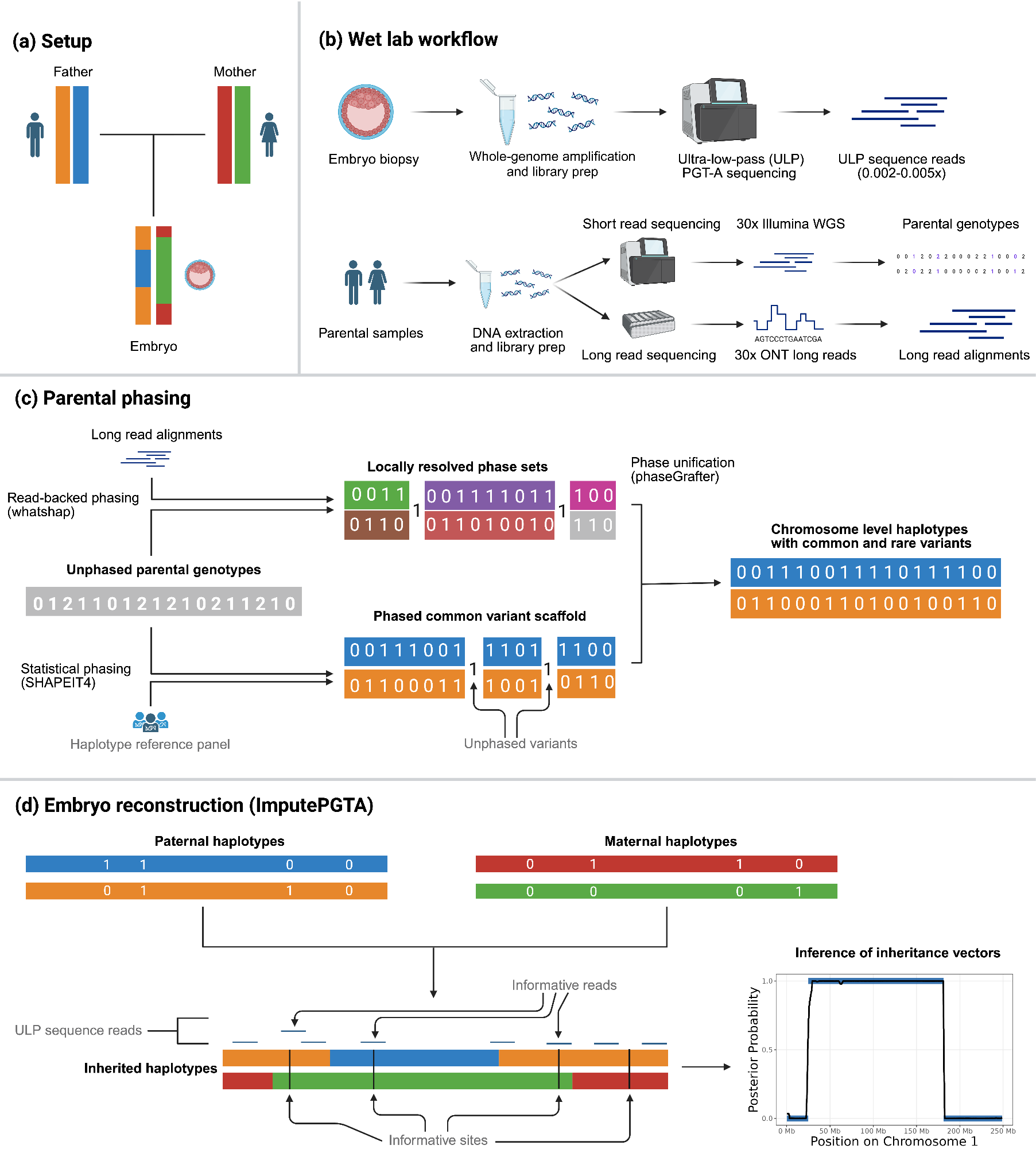
In order to perform reconstruction using ImputePGTA, however, we must first be able to accurately distinguish the two copies of each parent’s chromosomes via a process known as haplotype phasing. It is not typically possible to produce sufficiently accurate haplotypes from the most common types of genome-wide data: short read sequencing and genotyping arrays. This is because using standard methods for phase estimation results in a proliferation of switch errors, which are errors in the estimated haplotypes where the phasing algorithm misassigns segments. These switch errors impact reconstruction in the same way as recombination events, which must similarly be identified by ImputePGTA from the ultra-sparse embryo data, which increases the difficulty of accurate reconstruction.
Although not necessary to perform embryo genome reconstruction with suitable accuracy for PGT-P in many cases, we can improve parental phasing by sequencing parents with long reads, which intrinsically carry phase information and thereby substantially increase the accuracy of haplotype estimates (Figure 2B). When data from grandparents are available, it can further improve phasing. In order to provide ImputePGTA with the most accurate parental haplotypes possible, we developed an end-to-end pipeline that integrates short and long read data from the parents (and grandparents, when available) to produce a final set of parental haplotypes that draws on all available information (Figure 2C).
ImputePGTA then takes these parental haplotype estimates along with the ultra-sparse embryo PGT-A data, identifies the locations of recombinations and switch errors, and infers the inherited genome of a given embryo at all sites where a parent has data (Figure 2D). We can then compute polygenic scores from the reconstructed embryo genome. Importantly, ImputePGTA produces well-calibrated posterior distributions of both the imputed embryo genotypes, as well as the polygenic scores, which allows us to appropriately propagate uncertainty in the reconstruction downstream to disease risk and trait prediction.
Validation
In order to ensure that our reconstruction algorithm worked both in theory and in practice, we validated ImputePGTA through extensive simulations, application to downsampled real data, as well as in six real-world cases across four families that underwent PGT-A testing and subsequently resulted in born children.
We began with gold-standard data from the Platinum Pedigree, a publicly available four-generation family often used to benchmark genomic methods (Kronenberg et al. 2024). We used the phased haplotypes from two parents in the dataset and simulated the genomes of five possible offspring of these parents using realistic empirical estimates of recombination rates (Bhérer et al., 2017).
We then simulated ultra-low coverage sequence data from these offspring at a wide range of coverages including those typical of PGT-A. To mimic the kinds of imperfections seen in real data, we also introduced varying levels of switch errors. These results told us that the accuracy of reconstruction increases with increasing amounts of data on the “embryos” and with decreasing amounts of error in the parental haplotypes used for reconstruction. Importantly, we found that even with coverages typical of ultra-sparse PGT-A data, it is possible to reconstruct and score the offspring genomes with accuracy comparable to specialized, purpose-built PGT-P assays.
The Platinum Pedigree also includes eight real offspring, five of whom have publicly available high-coverage short-read sequencing data. This allowed us to test the accuracy of our reconstructions on real sequence data, by comparing the imputed genotypes from our algorithm to the known, true genotypes of the children. These results followed the same qualitative patterns as in the simulations, thus verifying that ImputePGTA “survived contact with reality” (i.e., empirical data).
We analyzed theoretically the loss in efficacy of screening based on PGT-A reconstructed embryo genomes compared to the true embryo genome, finding a loss of only 5-10%, provided that the statistical phasing quality of parents was good.
The ultimate test, however, is to validate whether this approach works in clinical contexts. To this end, we received IRB approval for a validation study in real families. We recruited four couples who had previously undergone IVF and had existing PGT-A data from different providers on at least one embryo, which was implanted and resulted in a successful pregnancy. With the help of the parents, we obtained the original PGT-A data used to screen these embryos, and we also obtained DNA samples from the born child corresponding with the screened embryo. In total, we obtained PGT-A data for six embryos/children across four families.
Each couple provided DNA samples, allowing us to perform comprehensive sequencing of both parents using both long and short reads. In one case, we also obtained and sequenced DNA from two of the four grandparents to further improve the accuracy of parental phasing.
With these inputs for ImputePGTA, we had the ingredients to apply our approach to reconstructing the embryo genome from PGT-A data. Then, with high-quality sequence data on the born child, we could compare the accuracy of the reconstruction to the known, true genotype of the same individual.
Results
Two of the six embryos were originally assayed using an ultra-low-pass sequence based PGT-A assay, while the other four were assayed using genotyping arrays, a legacy and non-standard technology that modern PGT-A providers have moved away from. In general, genotyping arrays provide denser information about embryos (and are therefore very easy to reconstruct whole genomes from in the ImputePGTA framework), but their greater expense has led to reduced use of this technology. As such, we focus on the results for the two ULP embryos (the results for the array cases can be found in the whitepaper for the interested reader).
For the two embryos tested with ultra-low-pass sequencing, ImputePGTA accurately reconstructed their genetic profiles. When evaluating genetic variants that differ between the parents—and therefore vary among siblings—ImputePGTA achieved an accuracy (dosage correlation) of 0.96 when using short and long reads for parental phasing. The polygenic scores calculated from these reconstructed genomes also closely matched the actual scores from the born children, with an average error (mean absolute error) of just 0.16 standard deviations (Figure 3). In our whitepaper, we further show that these polygenic scores, which are the primary focus in embryo screening, match the accuracy of scores typically used in academic genomic research.
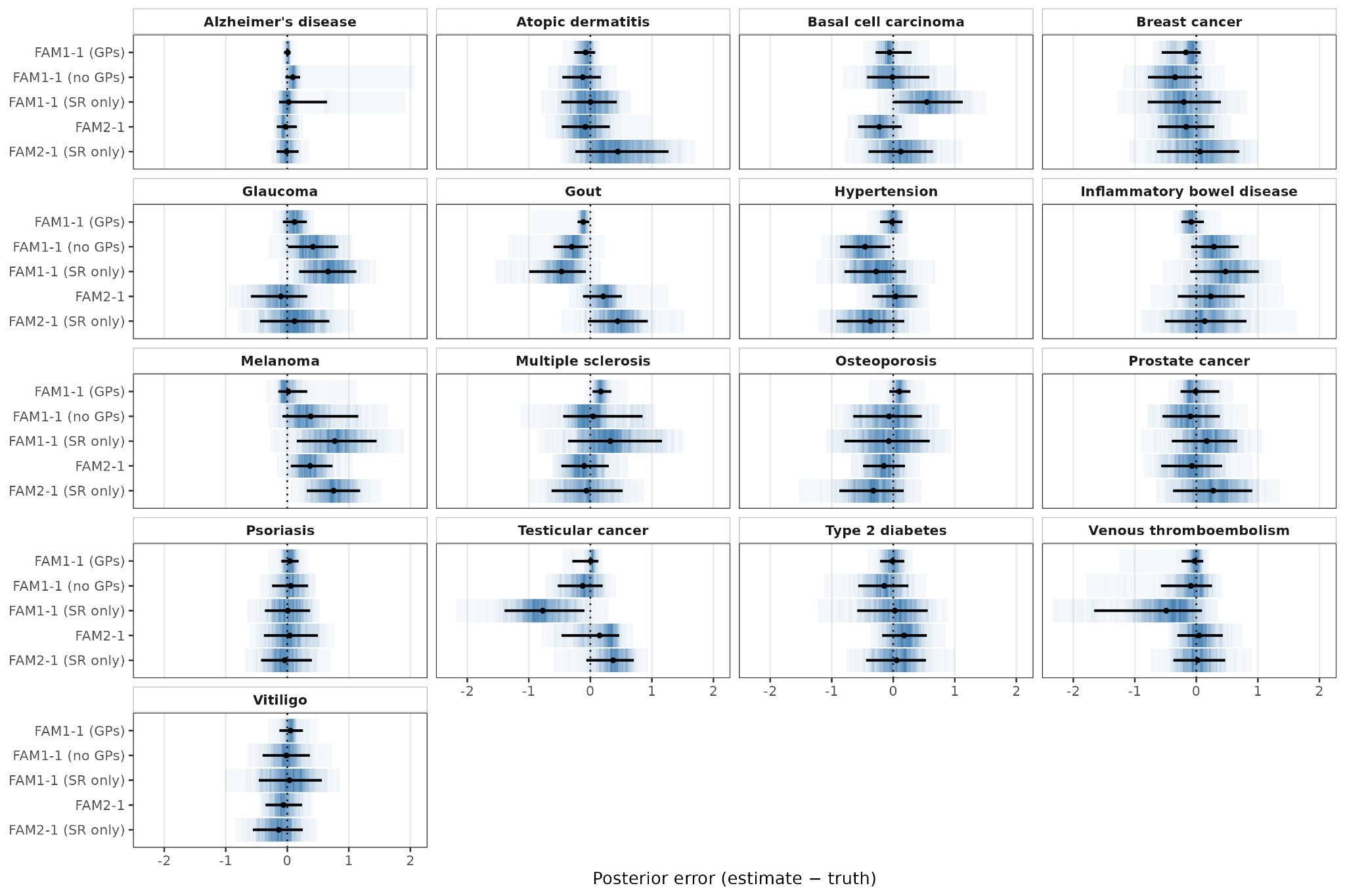
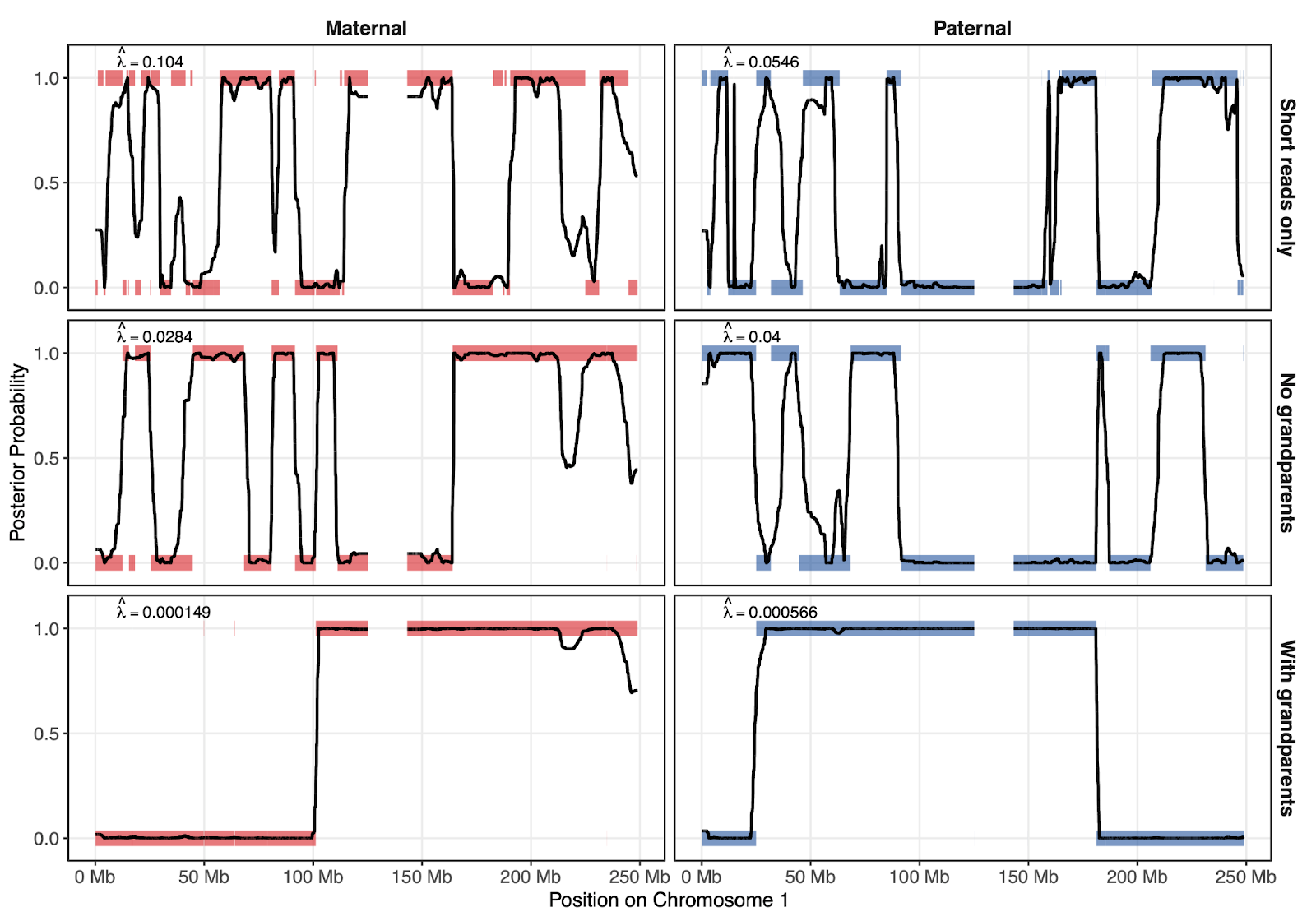
Figure 4 shows how parental phasing quality affects the reconstruction of the embryo genome as a mosaic of the parents’ haplotypes. Here, the y axis represents which parental haplotype was copied from (either 0 or 1). The shaded colored lines represent the true inheritance pattern, and the black lines represent the inheritance pattern that ImputePGTA inferred. The closer the black lines track the shaded bars, the more accurately the embryo genotypes were reconstructed. Intermediate values of the black line between 0 and 1 represent regions of greater uncertainty, where reconstruction is expected to perform more poorly.
Each successive row in the figure depicts the results as we include more information that helps phase the parents – the top row is when we only phase using short reads, the middle when we phase with both short and long reads, and the bottom row when we phase with short and long reads and with grandparental data.
The better the parental phasing is, the less fragmented the true inheritance pattern of the embryos appears, and the better the reconstruction is. For example, the dosage correlation (a measure of accuracy) of the embryo reconstruction increases from 0.92 when using only short reads, to 0.96 when using both short and long reads, to 0.99 when we use short reads, long reads, and grandparental data.
Notably, ImputePGTA doesn’t just work for polygenic traits, where thousands of genetic variants collectively influence a score; by using long reads it can also pinpoint critical single variants of clinical importance: In our validation study, we detected a rare pathogenic variant carried by the father in one family, which was inherited by the child.
Standard methods relying solely on short-read sequencing and phasing using haplotype reference panels would likely have missed this potentially vital finding due to its extremely low allele frequency (about one in 250,000). This example demonstrates the clinical value of integrating long-read sequencing for parental genomes, ensuring that even single, high-impact genetic variants are accurately captured.
Conclusion
ImputePGTA has enormous significance for parents using IVF. It makes polygenic screening much easier to access. But it also makes it imperative for companies like ours to try to educate physicians, parents, and professional associations on the science of polygenic prediction. We attempt to do this in our recent editorial for the American Society for Reproductive Medicine, in our publications, and in our meetings with clinicians and physicians.
We cannot eliminate disease risks entirely. But when it is done right, polygenic screening is a powerful way to stack the genetic deck in favor of conceiving children who live a healthy, happy, and long life. ImputePGTA helps significantly lower the barrier to entry for parents wishing to achieve these goals.
—
If you are interested in learning more, please take a look at our ImputePGTA whitepaper. Subscribe to our newsletter for more updates.

Do I understand correctly in the top of the page poster, that Embryo 3 is optimal for all three phenotypes: IQ 126, Alzheimer's risk 1.8% and T2D risk 11%? If so, it makes sense: T2D is known to accelerate Alzheimer's and higher IQ correlates with lower risk of both diseases.
Is this what the example poster intends to portray?