

ImputePGTA 2.0: Polygenic Embryo Screening for Every Ancestry
Our new algorithm closes the ancestry gap in embryo screening by jointly modeling all embryos in an IVF cycle

In November, we announced ImputePGTA, a technique that enables accurate polygenic embryo screening from routine PGT-A data already generated at IVF clinics worldwide. Today, we are announcing ImputePGTA V2, which substantially improves accuracy through a novel algorithm that jointly models and estimates genotypes for all available embryos, with the biggest gains for parents from underrepresented ancestries.
To appreciate this upgrade, it is worth revisiting the core mechanic of ImputePGTA. The algorithm treats the embryo as a mosaic of its parents. By combining the ultra-sparse sequencing from a standard aneuploidy test with high-coverage sequencing of the parents, we can mathematically impute the missing pieces of the embryo’s genome. This allows us to reconstruct a complete genetic profile and calculate polygenic scores for each embryo using only the sparse data already collected by IVF clinics.
However, the accuracy of this reconstruction relies on our ability to accurately map the parents’ genomes first. And this brings us to one of the most persistent challenges in modern genetics.
Table of Contents
The ancestry problem in genomics
The reference panels, genome-wide association studies, and computational tools that underpin modern genetic prediction were overwhelmingly developed using data from people of European descent. This has practical consequences for anyone whose ancestry falls outside that narrow window.
One place this shows up is in haplotype phasing, the process of determining which genetic variants sit together on the same copy of a chromosome. Humans carry two copies of each chromosome, one from each parent. Phasing is the process of figuring out which mutations belong together on the same inherited strand. Accurate phasing is essential for ImputePGTA, because our algorithm reconstructs the embryo genome by figuring out which segments of each parent’s chromosomes were passed down to the embryo. If we cannot accurately distinguish the two copies of a parent’s chromosomes in the first place, the accuracy of the reconstruction suffers.
The problem is that phasing quality depends heavily on the reference panels available for a given ancestry. For parents of European descent, where reference panels are large and well-curated, statistical phasing tends to be quite accurate. For parents of East Asian, African, South Asian, or other ancestries currently underrepresented in biobanks, phasing produces substantially more errors, known as switch errors, where segments of the two chromosome copies are incorrectly swapped.
To illustrate: in our validation study, the European-ancestry parents we analyzed had estimated switch error rates (λ) of 0.07–0.21 per centimorgan when phased using standard whole-genome sequencing, corresponding roughly to one incorrect phase switch every several megabases of DNA. The East Asian mother in one of our families (FAM6) had an estimated switch error rate of 1.69 per centimorgan, roughly 10 to 20 times worse. This translates directly into more difficult and less accurate embryo genome reconstruction.
In V1 of ImputePGTA, which processed each embryo independently, these switch errors directly degraded reconstruction accuracy. The algorithm had to identify true recombination events (where the embryo actually inherited a different parental segment) and artifacts of phasing error, and it had to do this from ultra-sparse PGT-A data alone. When switch errors were frequent, this became much harder, creating a real disparity in the quality of results parents could expect depending on their ancestry. In such circumstances, phasing quality could only be substantially improved by sequencing the grandparents of the embryos. However, this approach is often time-consuming, logistically complex, and in many cases impossible.
Joint modeling: using data from all embryos together
ImputePGTA V2 takes a fundamentally different approach from V1: it models all embryos from an IVF cycle simultaneously.
Why does this help? The key insight is that phasing errors leave a shared, recognizable signature when examined across multiple embryos. A true recombination event is unique to a single embryo. It happens during the formation of the egg or sperm that produced that specific embryo, and it will not appear in the same location in sibling embryos. A switch error in the parental haplotypes, on the other hand, is a property of the parents’ data, not the embryo’s. It generates a systematic pattern of apparent inheritance switches across every embryo that inherited that chromosomal segment.
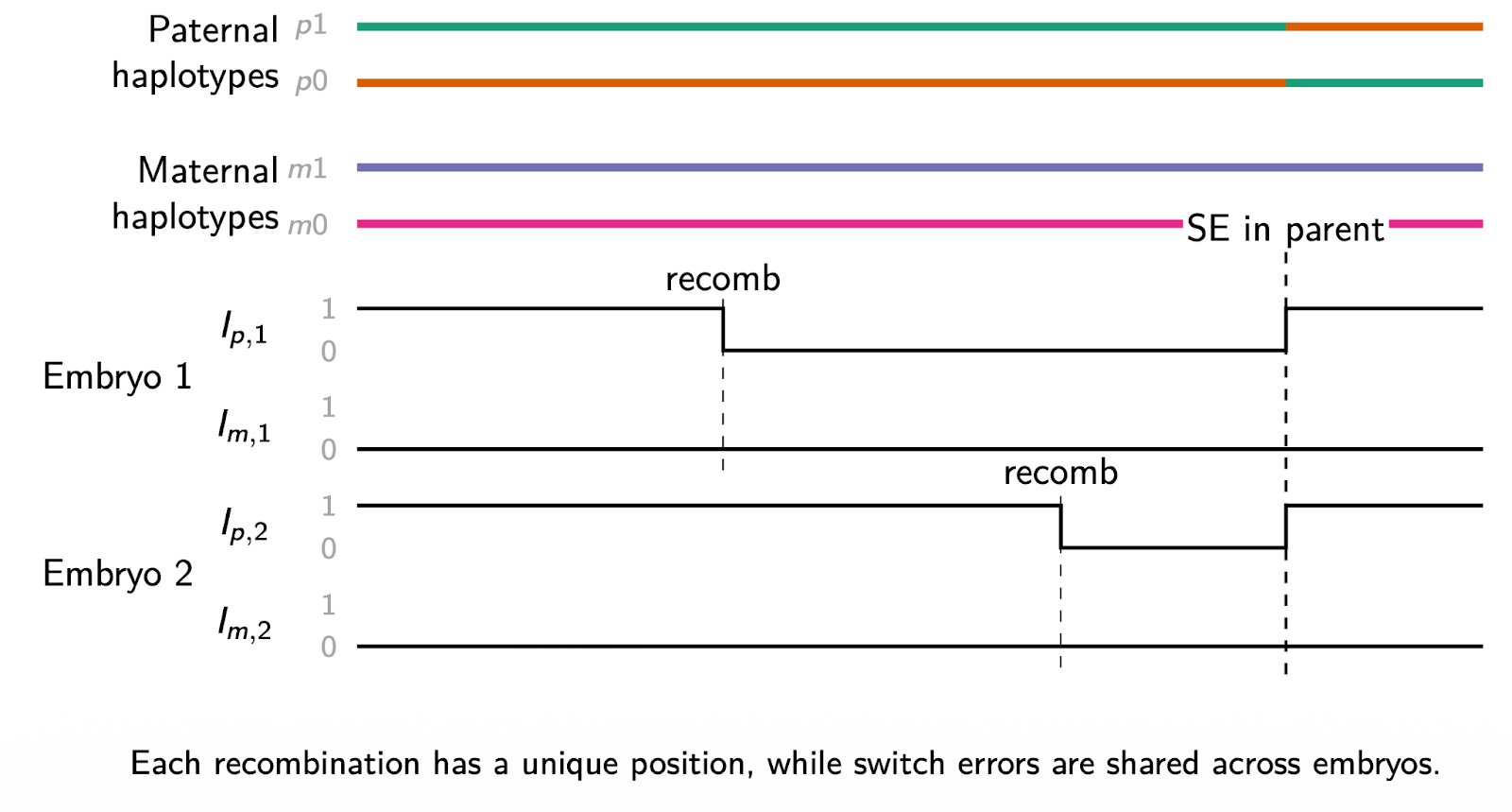
By examining multiple embryos jointly, V2 can distinguish these two signatures and directly correct for phasing errors. The more embryos available, the more statistical power the algorithm has to detect and correct switch errors.
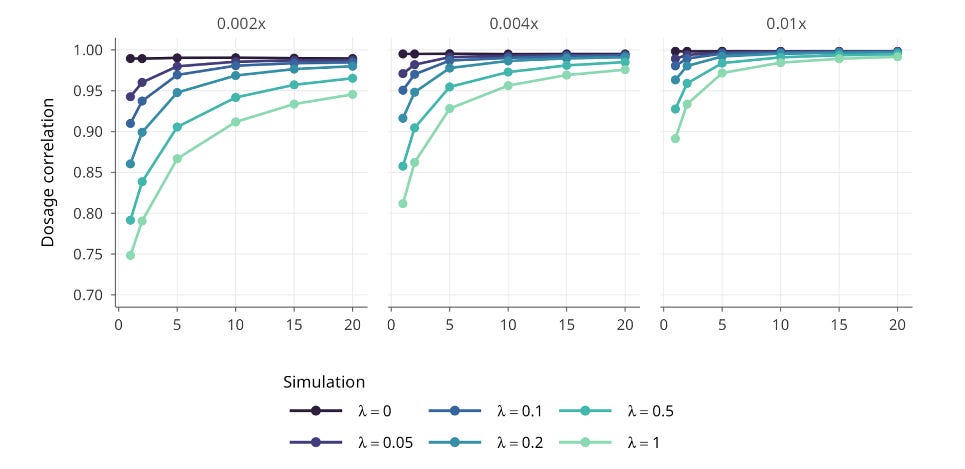
A typical IVF cycle for a 30-year-old woman yields around 8 total embryos (see our previous analysis of IVF yields), which provides substantial information for joint modeling. In simulations under the most challenging conditions, with high switch-error rates and ultra-sparse sequencing, jointly modeling just five embryos preserved about 85% of ideal selection performance. With better statistical phasing or more embryos, the loss shrinks to roughly 2 to 3%.
To put that in perspective: the performance loss from using standard genotyping arrays in academic research, where missing variants are statistically inferred from reference panels, is larger than the loss we observe using ultra-sparse PGT-A data with joint modeling.
The technical challenge
Joint modeling of embryos is conceptually simple but computationally extremely difficult. It requires keeping track of which copy of each parent’s chromosome every embryo inherited, along the length of the genome. In technical terms, this generalizes a classic inheritance-tracking method known as the Lander–Green algorithm.
The problem is that the complexity of the Lander-Green model grows exponentially with the number of embryos. Because the algorithm must simultaneously track which parental haplotype was inherited at each position across all embryos and correct for errors in the parents, the complexity explodes. For example, naively modeling 8 embryos would require tracking 2^16 = 65,536 possible inheritance states at every genomic position, which is already close to computational intractability, and this grows to over a million states for 10 embryos.
We developed a more efficient inference method that avoids this combinatorial explosion. Instead of tracking every possible inheritance pattern, the algorithm pools information across siblings to progressively refine the parental haplotypes. In practice, this allows us to model dozens of embryos simultaneously without prohibitive computational cost.
Every embryo counts, even aneuploid ones
In any IVF cycle, a significant proportion of embryos are aneuploid, meaning they have chromosomal abnormalities such as extra or missing chromosomes. The rate of aneuploidy increases with maternal age: for women over 40, the majority of embryos may be aneuploid. These embryos are not candidates for transfer and will not result in a pregnancy.
For many parents, learning that most of their embryos are aneuploid is one of the most difficult moments in the IVF process. But V2 introduces an unexpected silver lining.
Aneuploid embryos still carry useful genetic information on their euploid (normal) chromosomes. An embryo with trisomy 21, for example, carries three copies of chromosome 21, while the remaining 21 autosomal pairs are typically euploid and informative. ImputePGTA can now incorporate data from these euploid chromosomes into the joint model, using the aneuploid embryos to help improve phasing correction and reconstruction accuracy for the euploid embryos that are actually being screened.
This means that in a cycle where a couple produces 8 embryos but only 3 are euploid, the remaining 5 aneuploid embryos are not wasted data. They actively contribute to more accurate screening of the viable embryos. In cycles with many aneuploid embryos, which is precisely when parents need the most accurate information about their remaining options, this additional data source can make a meaningful difference.
Validation across diverse ancestries
We validated ImputePGTA V2 in several families spanning multiple ancestries, including European, East Asian, South Asian, and admixed (European/African/Amerindian) backgrounds.
For three families with ultra-low-pass PGT-A sequencing data and born-child validation data (FAM1, FAM2, FAM5), the average dosage correlation between imputed and true genotypes at informative sites was 0.980 when using both short and long reads for parental phasing. The polygenic scores calculated from these reconstructed genomes closely matched the actual scores from the born children, with an average error of just 0.11 standard deviations across 17 disease traits.
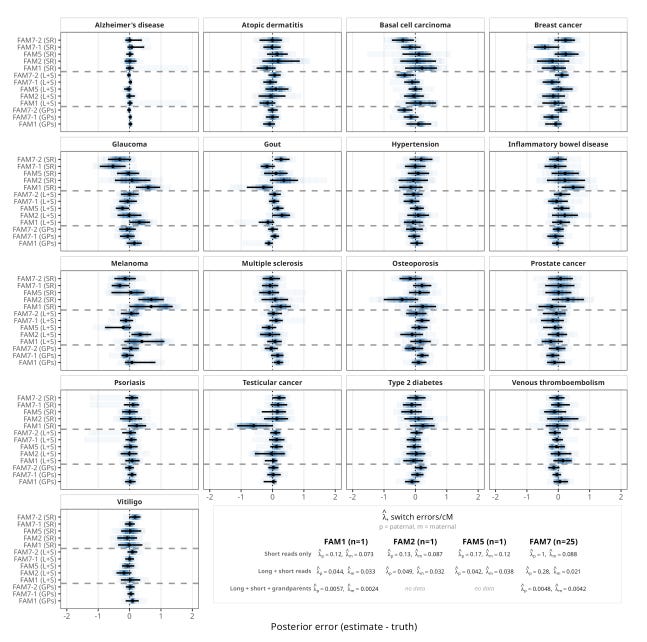
We also validated ImputePGTA V2 in a seventh family (FAM7) with admixed South Asian and East Asian ancestry, for which we had high-coverage whole-genome sequencing data on 25 embryos. This family provides the clearest demonstration of joint modeling’s power. Using only short-read parental phasing with the V1 model (single-embryo inference), the average dosage correlation was 0.835. When all 25 embryos were jointly modeled using V2, this rose to 0.966, a dramatic improvement driven entirely by the algorithm’s ability to leverage data from all the embryos to detect and correct parental switch errors.
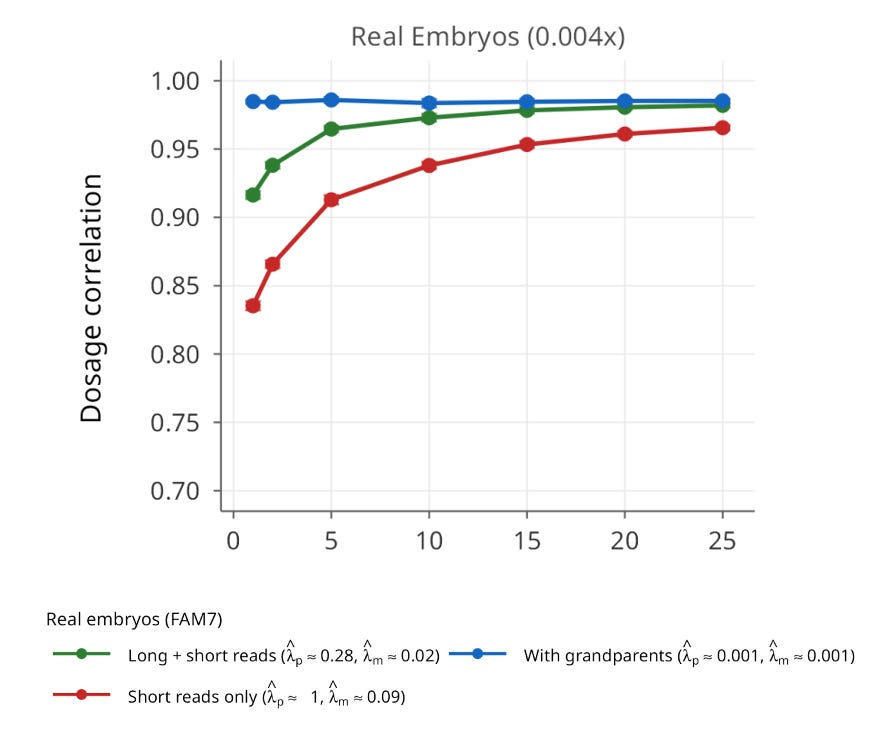
Joint modeling has a useful side effect: while inferring the embryos’ genomes, it also detects and corrects switch errors in the parental haplotypes, producing more accurate parental maps in the process. Those refined maps can then support more reliable ancestry estimates and relatedness analyses.
What this means for parents
When we announced ImputePGTA V1 in November, we described it as a technology that “brings polygenic embryo prediction to IVF clinics worldwide.” V2 makes that promise more concrete than ever. The worse the phasing, the more V2 helps, which means the families who previously stood to benefit least now see the largest improvements.
A couple in Seoul, Lagos, Dubai, or Mumbai using a local IVF clinic that offers PGT-A can now access polygenic screening at a quality level that approaches what was previously only achievable for European-ancestry families with the best available data. Every embryo in their cycle, including the aneuploid ones, contributes to better results. The algorithm scales to handle as many embryos as are available, and the barrier to entry continues to drop.
Polygenic screening cannot eliminate disease risk. But when it is done right, it is a powerful way to give children the best possible genetic start in life. ImputePGTA V2 ensures that this opportunity is not limited by ancestry or geography.
For full technical details, see our ImputePGTA V2 preprint. For background on the original ImputePGTA method, see our previous Substack post. If you are interested in polygenic embryo screening for your IVF cycle, sign up here.
Subscribe to our newsletter for more updates.

Really smart approach to the ancestry equity problem. Using aneuploid embryos as informative datapoints for phasing correction is genuinely clever, kinda like turning waste into signal. I've seen similar patterns in distributed systems where combining weak signals outperforms individual strong ones under the right conditions.